Buscando, por favor espere.
 Imprimir
Imprimir
 Añadir a biblioteca
Añadir a biblioteca
 Comentar este artículo
Comentar este artículo
Molina Arias M. Principales algoritmos de aprendizaje automático. Parte 2. Evid Pediatr. 2026;22:20.
En el artículo anterior de esta serie de Fundamentos de Medicina Basada en la Evidencia analizamos algunos de los algoritmos de aprendizaje automático supervisado más clásicos: la regresión lineal, la regresión logística y las máquinas con vectores de soporte. En esta segunda entrada, completaremos el bloque del aprendizaje automático supervisado, abordando herramientas de gran utilidad y potencia en la investigación biomédica, tales como los modelos basados en árboles de decisión (tanto simples como combinados) y los clasificadores basados en probabilidad y proximidad (el algoritmo naive-Bayes y el de los k vecinos más cercanos).
Por último, en un tercer artículo nos centraremos en los algoritmos de aprendizaje no supervisado, orientados al descubrimiento de patrones sin necesidad de etiquetas previas durante su fase de entrenamiento.
El árbol de decisión simple es un algoritmo de aprendizaje supervisado que se utiliza para predecir o clasificar una variable objetivo basándose en una serie más o menos larga de variables predictoras o independientes.
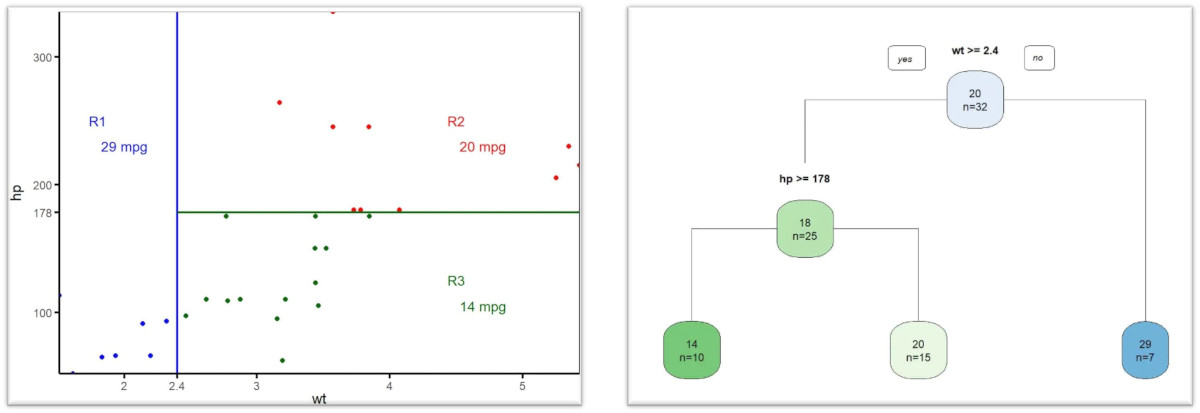
Su objetivo primordial es estratificar el espacio de las variables predictoras en regiones diferenciadas y no solapadas para realizar predicciones ante posibles datos nuevos, cuyo valor de la variable objetivo será el de la media de la región que le corresponda, en casos de regresión, o el de la categoría más frecuente, en el caso de árboles de clasificación (Figura 1).
Figura 1. Estratificación del espacio de variables predictoras y su representación gráfica final. Mostrar/ocultar
En la práctica, este proceso se muestra de forma gráfica como un árbol invertido (las raíces en la parte superior y las hojas en la inferior), donde cada nodo representa una variable que permite tomar una decisión sobre la bifurcación a una de estas áreas, en función del valor de esa variable. Finalmente, cada nodo terminal (hoja) representa el resultado de la decisión o la clasificación final.
Estos modelos son especialmente útiles cuando los datos siguen relaciones no lineales complejas, situación en la que pueden superar en precisión a los modelos lineales tradicionales, como la regresión lineal o logística múltiple.
Ya hemos dicho que el funcionamiento del algoritmo se basa en la división del espacio multidimensional de las variables predictoras en una serie de zonas o regiones de decisión, asignando a cada nueva observación el valor medio de su región correspondiente. Para definir matemáticamente estas particiones, durante el entrenamiento el algoritmo busca minimizar el error utilizando el error cuadrático medio como función de coste, calculando la diferencia entre los valores observados y los predichos (recordemos que, al tratarse de aprendizaje supervisado, conocemos el valor de la variable objetivo durante el entrenamiento). Este método es similar al que vimos para el entrenamiento de la regresión lineal.
Dado que evaluar exhaustivamente todas las combinaciones posibles de particiones puede resultar computacionalmente inviable, el algoritmo emplea un enfoque ávido o voraz (greedy approach) de diseño descendente. Mediante un proceso de división binaria recursiva, el modelo toma decisiones localmente óptimas en cada iteración.
Así, en cada nodo evalúa todos los predictores y selecciona la división que genere la mayor reducción inmediata del error cuadrático medio en ese nodo específico. Este método prioriza la optimización a corto plazo sin proyectar su impacto en los niveles inferiores del árbol, repitiendo el ciclo de partición hasta alcanzar los criterios de parada predefinidos (por ejemplo, alcanzar un número mínimo de elementos por nodo).
El problema es que este método puede proporcionar buenas predicciones a costa de aumentar la complejidad del árbol (lo que disminuye su interpretabilidad) y de caer en el sobreajuste (disminuyendo su validez externa). Para controlar este inconveniente, existen una serie de técnicas o alternativas.
La primera opción es limitar el crecimiento del árbol desde el inicio (pre-poda), estableciendo criterios de parada predeterminados, como un número máximo de nodos, un mínimo de elementos por hoja o un umbral límite para la reducción del cuadrado de los residuos.
Una alternativa más robusta es permitir que el árbol crezca completamente y luego aplicar un proceso de simplificación conocido como poda (pruning).
Otra posibilidad es emplear una técnica de poda basada en la complejidad aplicando una penalización a la función de mínimos cuadrados, de forma análoga a la regularización lasso que vimos al hablar de regresión. Este método penaliza la función de coste original, sumándole un factor proporcional al número de nodos terminales del árbol, ponderado por un hiperparámetro de regularización (α), cuyo valor debe definir el investigador. Valores más elevados de este hiperparámetro fuerzan la eliminación progresiva de las ramas que aportan menos poder predictivo.
Estos algoritmos siguen un razonamiento similar al de los árboles de regresión, pero tienen algunas diferencias, sobre todo en la función de coste que se utiliza para construir el árbol, ya que requieren métricas específicas diseñadas para evaluar la homogeneidad cualitativa dentro de cada región o nodo.
Lo más intuitivo sería emplear la tasa de error de clasificación (la proporción de elementos mal clasificados en cada nodo) como función de coste, pero carece de la sensibilidad matemática necesaria para guiar el crecimiento de un modelo preciso. Por ello, su uso suele limitarse a la fase de poda de un árbol ya construido. En su lugar, los algoritmos recurren a dos índices matemáticos diferentes.
El índice de Gini cuantifica la varianza total entre las diferentes categorías. Se considera la medida estándar de la pureza del nodo; un valor de Gini cercano a cero indica que la inmensa mayoría de las observaciones en ese nodo pertenecen a una única clase diagnóstica. El índice de entropía, inspirado en la teoría de la información, mide el grado de incertidumbre o “desorden” en la distribución de los datos. El algoritmo busca dividir las variables de forma que se minimice esta entropía, lo que equivale a maximizar la homogeneidad categórica.
Dado que tanto el índice de Gini como la entropía persiguen el mismo objetivo (maximizar la pureza diagnóstica de los nodos terminales), en la práctica arrojan resultados sumamente similares.
Los hiperparámetros habituales que el investigador debe ajustar son:
En cuanto a las métricas de desempeño, son las habituales que vimos al hablar de otros algoritmos de aprendizaje supervisado. En el caso de árboles de clasificación se recurre al error cuadrático medio, al error absoluto medio o al coeficiente de determinación. En el caso de los árboles de regresión, son las habituales de pruebas diagnósticas, aunque ya vimos que en ciencia de datos suelen valorarse preferentemente sensibilidad, valor predictivo positivo, F1-score y curvas ROC.
El rendimiento de los árboles de decisión frente a otros algoritmos o métodos estadísticos clásicos depende de la naturaleza del problema. Si el objetivo es predecir una variable continua y los datos se ajustan a un modelo lineal, la regresión tradicional ofrecerá mejores resultados. Sin embargo, si los datos presentan relaciones no lineales y complejas, los árboles de decisión suelen superar a los modelos lineales al capturar mejor estas interacciones.
La indudable ventaja de un árbol simple es su alta interpretabilidad clínica y su fácil visualización gráfica. No obstante, presentan limitaciones notables: generalmente no alcanzan niveles de precisión tan altos como otros algoritmos y carecen de robustez.
Esta falta de robustez se traduce en una alta varianza; es decir, pequeñas variaciones en los datos de entrenamiento pueden alterar drásticamente la estructura topológica del árbol construido, lo que los hace muy propensos al sobreajuste. Para tratar de soslayar estos inconvenientes, se han desarrollado los algoritmos de árboles de decisión combinados, que veremos a continuación.
Los árboles de decisión muestran claramente el esfuerzo continuo para optimizar el denominado compromiso sesgo-varianza. El sesgo representa el error sistemático derivado de sobresimplificar un problema (conduciendo al subajuste), mientras que la varianza refleja la sensibilidad excesiva del modelo al ruido específico de los datos de entrenamiento (conduciendo al sobreajuste).
Dado que los árboles de decisión simples son estructuralmente inestables y presentan una alta varianza (riesgo de sobreajuste), se han desarrollado métodos de ensamblaje (ensemble) que combinan múltiples árboles para generar un modelo final significativamente más robusto y preciso, capaz de generalizar sus predicciones con nuevos datos.
El término bagging deriva de bootstrap aggregation. Esta metodología reduce la varianza construyendo múltiples árboles de decisión independientes. Para lograrlo ante un único conjunto de datos, el algoritmo genera múltiples submuestras mediante técnicas de bootstrapping (remuestreo aleatorio con reemplazo). A continuación, se entrena un árbol completo en cada submuestra. La predicción final se obtiene promediando los resultados de todos los árboles (en problemas de regresión) o mediante votación mayoritaria (en clasificación).
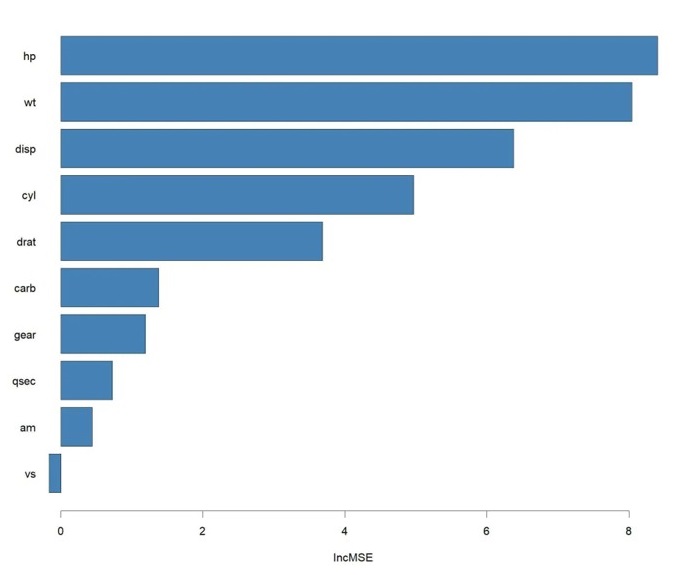
Al combinar cientos de árboles, se sacrifica la interpretabilidad visual directa del modelo: ya no podemos representar el árbol gráficamente. Para compensar esta pérdida de interpretabilidad, se evalúa la importancia de las variables. El algoritmo cuantifica matemáticamente cuánto aumenta el error del modelo (mínimos cuadrados en regresión o Gini en clasificación) al permutar o excluir cada variable predictora, permitiendo al investigador identificar qué factores clínicos determinan realmente la predicción (Figura 2).
Figura 2. Representación gráfica de la importancia de las variables predictoras utilizadas en la elaboración de árboles de decisión combinados. Se muestran, en orden descendente, las variables con mayor participación en la capacidad predictiva del modelo. Mostrar/ocultar
El bagging tradicional presenta una limitación metodológica: si existe una variable clínica con un poder predictivo abrumadoramente superior al resto, la inmensa mayoría de los árboles generados la seleccionarán para su partición inicial. Esto produce un “bosque” de árboles estructuralmente muy similares y altamente correlacionados, lo que limita la reducción de la varianza.
Los bosques aleatorios mitigan esta autocorrelación modificando la regla de partición. Mientras que la técnica de bagging evalúa todas las variables en cada nodo, el bosque aleatorio tiene restringida esta evaluación de todas las variables disponibles y solo se le permite considerar un subconjunto aleatorio de predictores (generalmente, la raíz cuadrada del número total de variables). Esto fuerza la diversidad estructural de los árboles, maximizando la robustez general del ensamble.
Mientras que el bagging y los random forest entrenan árboles de forma simultánea e independiente, las técnicas de boosting operan de manera secuencial. El proceso comienza con un árbol sumamente simple (con alto sesgo). Posteriormente, se añaden nuevos árboles de forma sucesiva, donde cada nuevo modelo se entrena específicamente para aprender y corregir los errores residuales cometidos por la secuencia de árboles que le preceden.
Esta técnica iterativa es excepcionalmente potente, ya que es capaz de reducir tanto el sesgo como la varianza simultáneamente. Esta familia de algoritmos es una de las más eficaces en el aprendizaje automático moderno, incluyendo implementaciones avanzadas como el Gradient Boosting, el XGBoost (que integra técnicas de regularización para evitar el sobreajuste), o variantes optimizadas como LightGBM y CatBoost.
En el espectro más avanzado, los algoritmos BART aplican un enfoque de modelado estadístico no paramétrico que fusiona la arquitectura de los árboles de decisión con los principios bayesianos. Además de su alta capacidad para modelar relaciones biológicas complejas y no lineales, su principal ventaja clínica es la provisión de inferencias probabilísticas, permitiendo al investigador no solo obtener una predicción, sino cuantificar la incertidumbre estadística que la acompaña.
El clasificador naive-Bayes (Bayes ingenuo) es un modelo probabilístico fundamentado directamente en el teorema de Bayes. Se denomina “ingenuo” porque asume, de forma estricta, que el valor de una variable predictora es matemáticamente independiente de las demás para una categoría diagnóstica concreta, supuesto que no siempre se cumple o es posible comprobar.
Aunque en la biología humana las variables clínicas casi siempre guardan cierta correlación, esta suposición simplifica radicalmente la complejidad del cálculo introduciendo un sesgo habitualmente aceptable. Es un algoritmo extremadamente rápido, que maneja muy bien espacios de alta dimensionalidad y con buenas predicciones incluso cuando se entrena con conjuntos de datos relativamente pequeños.
Existen varios tipos de este algoritmo, que varían en su metodología, en los hiperparámetros que utilizan y en el tipo de variables predictoras más convenientes para su funcionamiento. El método gaussiano, que se utiliza cuando las variables predictoras son continuas y siguen una distribución normal, tiene como hiperparámetros la varianza y un término de suavizado que es útil cuando hay problemas con probabilidad cero.
El método multinomial es útil para el manejo de datos discretos, como es el caso de las frecuencias de palabras en la clasificación de texto. Utiliza un parámetro de suavizado de Laplace (α), especialmente útil con datos escasos. Por último, el método naive-Bayes Bernouilli se utiliza con datos binarios. Además del parámetro de Laplace, puede utilizar un parámetro de binarización del umbral, que convierte variables continuas en binarias.
El algoritmo k-nearest neighbors (k-NN) proporciona a posteriori la probabilidad de que un elemento pertenezca a una categoría en función de la categoría de los k elementos más cercanos en el espacio de las variables predictoras.
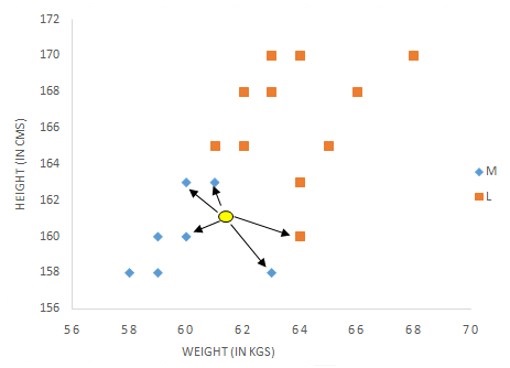
Su funcionamiento es bastante sencillo, aunque puede ser costoso desde el punto de vista computacional en espacios multidimensionales (Figura 3). En primer lugar, se representa el nuevo elemento del que se quiere hacer la predicción en este espacio multidimensional. A continuación, se calcula la distancia con los demás elementos y se seleccionan los k más próximos. La predicción será la categoría más frecuente entre estos k elementos, en los casos de clasificación, o la media o mediana de los mismos, en los casos de regresión. Para minimizar el error, durante la fase de entrenamiento se utiliza como función de coste la exactitud diagnóstica, para las predicciones categóricas, o el error cuadrático medio para las continuas.
Figura 3. Representación gráfica del algoritmo de los k vecinos más cercanos. Mostrar/ocultar
El principal hiperparámetro de este algoritmo es el número de vecinos (k), que suele calcularse empleando técnicas de validación cruzada. Un valor de k muy pequeño favorecerá el sobreajuste y una mayor varianza: si cambiamos ligeramente el conjunto de entrenamiento, las predicciones del modelo cambiarán drásticamente. Por el contrario, el modelo será estable y robusto con un número k elevado, pero perderá flexibilidad y capacidad para capturar patrones sutiles (subajuste).
Otros hiperparámetros son la métrica de distancia utilizada (euclidiana, Manhattan, Minkowski, etc.) y el peso de los vecinos, que pueden ponderarse en función de la distancia al nuevo elemento.
Entre las ventajas del algoritmo k-NN están su simplicidad, su versatilidad para regresión y clasificación, el ser no paramétrico y el buen funcionamiento con conjuntos de datos pequeños y con clasificación multiclase.
Su mayor inconveniente es su sensibilidad a la escala de las variables predictoras (mide distancias), lo que obliga al preprocesamiento y estandarización de los datos, su sensibilidad al valor de k elegido y su tendencia a favorecer la clase más frecuente cuando las categorías están desbalanceadas.
Molina Arias M. Principales algoritmos de aprendizaje automático. Parte 2. Evid Pediatr. 2026;22:20.


 Artículo completo
Artículo completo
 PDF
PDF